
Craniosynostosis Craniofacial Anomaly
What is craniosynostosis?
Other conditions in which craniofacial anomalies may occur:
- Apert's Syndrome - The head usually appears short in the front and back and may be pointed at the top. Other characteristics may include webbed fingers and toes, widely spaced and protruding eyes, and dental crowding.
- Carpenter Syndrome - This syndrome typically includes traits such as abnormally short fingers, webbed toes, extra toes, underdeveloped jaw, highly arched palate, widely spaced eyes, and/or low-set, deformed ears. Half of patients with Carpenter syndrome also have heart defects.
- Cleft Palate / Cleft Lip - The incomplete closure of the lip and/or the roof of the mouth results in this defect.
- Crouzon's Syndrome - Characterized by abnormalities in the skull and facial bones, this syndrome often causes the skull to be short in the front and the back. Flat cheek bones and a flat nose are also typical of this disorder.
- Pfeiffer Syndrome - This rare disorder is characterized by abnormalities of the skull, hands, and feet.
- Saethre-Chotzen - An unusually short or broad head is usually an indicator of this disorder. In addition, the eyes may be spaced wide apart and have droopy eyelids, and fingers may be abnormally short and webbed.
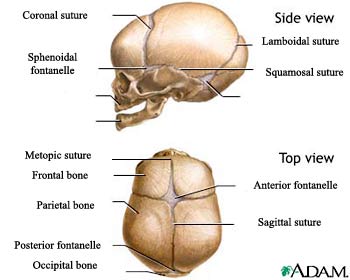
The normal skull consists of several plates of bone that are separated by sutures. The sutures (fibrous joints) are found between the bony plates in the head. As the infant grows and develops, the sutures close, forming a solid piece of bone, called the skull.
Craniosynostosis is a condition in which the sutures close too early, causing problems with normal brain and skull growth. Premature closure of the sutures may also cause the pressure inside of the head to increase and the skull or facial bones to change from a normal, symmetrical appearance.
What causes craniosynostosis?
Craniosynostosis occurs in one out of 2,000 live births and affects males twice as often as females. Craniosynostosis is most often sporadic (occurs by chance). In some families, craniosynostosis is inherited in one of two ways:
- Autosomal recessive - Autosomal recessive means that two copies of the gene are necessary to express the condition, one inherited from each parent, who are obligate carriers. Carrier parents have a one in four, or 25 percent, chance with each pregnancy, to have a child with craniosynostosis. Males and females are equally affected.
- Autosomal dominant - Autosomal dominant means that one gene is necessary to express the condition, and the gene is passed from parent to child with a 50/50 risk for each pregnancy. Males and females are equally affected.
Craniosynostosis is a feature of many different genetic syndromes that have a variety of inheritance patterns and chances for reoccurrence, depending on the specific syndrome present. It is important for the child as well as family members to be examined carefully for signs of a syndromic cause (inherited genetic disorder) of craniosynostosis such as limb defects, ear abnormalities, or cardiovascular malformations.
What are the different types of craniosynostosis?
There are numerous types of craniosynostosis. Different names are given to the various types, depending on which suture, or sutures, are involved, including the following:
Normal Skull of the Newborn
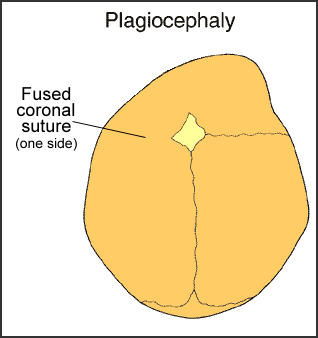
Plagiocephaly
Plagiocephaly occurs the most often. It happens in approximately one out of every 2,500 births. It involves fusion of either the right or left side of the coronal suture that runs from ear to ear. This is called coronal synostosis and it causes the normal forehead and the brow to stop growing. Therefore, it produces a flattening of the forehead and the brow on the affected side, with the forehead tending to be excessively prominent on the opposite side. The eye on the affected side may also have a different shape. There may also be flattening of the back area (occipital). Unilateral lambdoidal suture synostosis may cause plagiocephaly, as well.
Positional plagiocephaly is the most common cause of plagiocephaly. This is not caused by unilateral synostosis, but rather by sleeping in one position. The part of the skull that is dependent (in one position) tends to flatten out. Usually no intervention is needed.
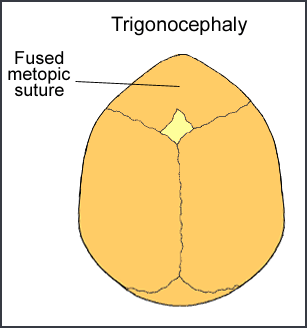
Trigonocephaly
Trigonocephaly is a fusion of the metopic (forehead) suture. This suture runs from the top of the head down the middle of the forehead, toward the nose. Early closure of this suture may result in a prominent ridge running down the forehead. Sometimes, the forehead looks quite pointed, like a triangle, with closely placed eyes (hypotelorism).
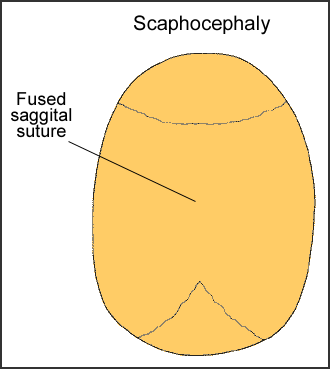
Scaphocephaly
Scaphocephaly is an early closure of fusion of the sagittal suture. This suture runs front to back, down the middle of the top of the head. This fusion causes a long, narrow skull. The skull is long from front to back and narrow from ear to ear.
What are the symptoms of craniosynostosis?
In infants with this condition, changes in the shape of the head and face may be noticeable. The appearance of the child's face may not be the same when compared to the other side. Other clinical findings may include the following:
- A full or bulging fontanelle (soft spot located on the top of the head)
- Sleepiness (or less alert than usual)
- Scalp veins may be very noticeable
- Increased irritability
- High-pitched cry
- Poor feeding
- Projectile vomiting
- Increasing head circumference
- Seizures
- Bulging eyes and an inability of the child to look upward with the head facing forward
- Developmental delays
The symptoms of craniosynostosis may resemble other conditions or medical problems. Always consult your child's physician for a diagnosis.
How is craniosynostosis diagnosed?
Craniosynostosis may be congenital (present at birth) or may be observed later, during a physical examination. The diagnosis is made after a thorough physical examination and after diagnostic testing. During the examination, your child's physician will obtain a complete prenatal and birth history of your child. He/she may ask if there is a family history of craniosynostosis or other head/face abnormalities. Your child's physician may also ask about developmental milestones since craniosynostosis can be associated with other neuromuscular disorders. Developmental delays may require further medical follow up for underlying problems.
During the examination, a measurement of the circumference of your child's head is taken and plotted on a graph to identify normal and abnormal ranges.
Diagnostic tests that may be performed to confirm the diagnosis of craniosynostosis include:
- X-rays of the head - a diagnostic test that uses invisible electromagnetic energy beams to produce images of internal tissues and bones of the head onto film.
- Computed tomography scan (also called a CT or CAT scan) of the head - a diagnostic imaging procedure that uses a combination of x-rays and computer technology to produce cross-sectional images (often called slices), both horizontally and vertically, of the head. A CT scan shows detailed images of any part of the body, including the bones, muscles, fat, and organs. CT scans are more detailed than general x-rays.
Treatment of craniosynostosis:
Specific treatment for craniosynostosis will be determined by your child's physician based on:
- Your child's age, overall health, and medical history
- Extent of the craniosynostosis
- Type of craniosynostosis (which sutures are involved)
- Your child's tolerance for specific medications, procedures, or therapies
- Expectations for the course of the craniosynostosis
- Your opinion or preference
Surgery is typically the recommended treatment. The goal of treatment is to reduce the pressure in the head and correct the deformities of the face and skull bones.
The optimal time to perform surgery is before the child is 1 year of age since the bones are still very soft and easy to work with. Surgery may be necessary at a much earlier age depending upon the severity of the condition.
Before surgery, your child's physician will explain the operation and may review "before and after" photographs of children who may have had a similar type of surgery.
Following the operation, it is common for the child to have a turban-like dressing around his/her head. The face and eyelids may be swollen after this type of surgery. The child is typically transferred to the intensive care unit (ICU) after the operation for close monitoring.
Problems after surgery may occur suddenly or over a period of time. The child may experience any or all of the following complications:
- Fever (greater that 101° F)
- Vomiting
- Headache
- Irritability
- Redness and swelling along the incision areas
- Decreased alertness
- Fatigue
These complications require prompt evaluation by your child's surgeon. The healthcare team educates the family after surgery on how to best care for their child at home, and outlines specific problems that require immediate medical attention.
Life-long considerations for a child with craniosynostosis:
The key to treating craniosynostosis is early detection and treatment. Some forms of craniosynostosis can affect the brain and development of a child. The degree of the problems is dependent on the severity of the craniosynostosis, the number of sutures that are fused, and the presence of brain or other organ system problems that could affect the child.
Genetic counseling may be recommended by the physician to evaluate the parents of the child for any hereditary disorders that may tend to run in families.
A child with craniosynostosis requires frequent medical evaluations to ensure that the skull, facial bones, and brain are developing normally. The medical team works with the child's family to provide education and guidance to improve the health and well being of the child.
Contact Us
For more information about UM Division of Plastic Surgery or to make an appointment, please call 1-800-492-5538 or 410-328-2360 (patients) or 1-800-373-4111 (physicians).